Pathogenetic Aspects of Infectious, Immunological, and Chronobiological Processes in Psychiatric Diseases
Henneberg AE, Kaschka WP (eds): Imunological Alterations in Psychiatric Diseases. Adv Biol Psychiatry, Basel, Karger, 1997, vol 18,pp 1-12.
VIRUSES AS ETIOLOGIC AGENTS OF SCHIZOPHRENIA
Robert H. Yolken, E. Fuller Torrey
Theoretical Background
Schizophrenia is a brain disease of unknown etiology which has a lifetime incidence of at least 1% of the population in North American and European countries. The occurrence of schizophrenia has been ascribed to numerous factors including psychodynamic, neurological, genetic, and environmental influences (1,2). There are several aspects of the epidemiology and pathophysiology of schizophrenia which suggests that infection with viruses or other microbial agents may play a role in the etiology of this disease (3). Evidence pointing to a role for viruses comes from several sources. Numerous studies have indicated that the season of birth is a major factor in determining whether an individual will develop schizophrenia during adult life. Generally, individuals born in the winter-spring months have the highest rate of disease, a timing consistent with the period of the year when transmissible infections are likely to occur (4). Furthermore, some, but not all, studies have reported an increased incidence of schizophrenia in children born during periods of increased infection with influenza virus in the population (5,6). The effect appears to be strongest when the possible viral exposure occurs during the third to seventh month of gestation (7,8). Additional studies have indicated that individuals who develop schizophrenia are more likely to be born during periods of increased activity of a number of infections, including measles, varicella zoster (chickenpox) and polio (9). Similarly, studies of birth order have indicated that individuals who are the product of later pregnancies are more likely to develop schizophrenia than first-born individuals, a finding consistent with the infection of the mother during pregnancy from young children at home (10). Studies of the prenatal and perinatal course of individuals who develop schizophrenia have also indicated that individuals who develop this disease are more likely to have a range of complications occurring during pregnancy, labor, or delivery (11,12). These findings are consistent with exposure to viruses or other environmental factors during pregnancy.
Epidemiological evidence for possible roles of viruses is supported by laboratory studies indicating that individuals with schizophrenia have antibodies to a range of viral agents in excess of that of control populations. Antibodies which have been identified include those directed at herpesviruses, encephalitis viruses, pestiviruses, and a range of other infections agents [reviewed in 3]. In some cases, the presence of antibodies to defined viral antigens correlates with alterations in brain structure as measured by neuroimaging analyses (13). A possible role of infectious agents if further supported by the recent development of animal models in which infections with a defined viral agent lead to persistent neurological infection associated with behavioral abnormalities. Of particular interest in this regard are animal models of brain infections with influenza virus (14), Borna virus (15), Sindbis virus (16), and the pestivirus bovine viral diarrhea virus (17). In the case of these models, infection of an animal early in life can lead to a range of neurological, behavioral, and systemic abnormalities which persist throughout the life of the animal. The expression of disease in these models following infection is dependent upon a number of genetic, immunological, and other host factors (tables 1, 2).
________________________________________________________________________________
Table 1: Mechanisms for chronic pathological changes
Persistent infection
DNA integration
Reverse transcription
Continuous replication
Cellular alteration
Cytolysis
Apoptosis
Membrane dysfunction
Immunopathology
Autoimmunity
T-cell dysfunction
Cytokine interactions
Neurotoxic metabolites
Table 2: Mechanisms for viral gene interaction
Immune dysfunction
T cell
B cell
NK cell
Macrophage
Cellular susceptibility
Cell surface receptors
Viral transporter proteins
Viral processing enzymes
Cytopathology
Oncogenes
Antioncognes
Apoptosis-associated proteins
Despite epidemiological and immunological evidence for viral infections, most studies have failed to consistently identify actively replicating viruses in samples from individuals with schizophrenia (18). There are several possible reasons for the inability to detect viruses in such samples. As noted above, epidemiological studies point to infections during the prenatal and early postnatal period as important factors in the development of schizophrenia. Similarly, prospective studies have identified neurological and cognitive abnormalities in children many years before the development of symptoms pathognomonic of schizophrenia (19). A patient with schizophrenia may thus present many years after the occurrence of the primary infection and the infecting agent may be difficult to detect at this point in the disease process. The recent development of methods for the detection of low levels of viral nucleic acids which may be present long after the occurrence of acute infections (20) may provide opportunities for the direct identification of antecedent viral infections in individuals presenting with schizophrenia.
There are several mechanisms by which the acute infection with a viral agent during the perinatal period can lead to a chronic disease process occurring later in life. For example, many viruses can establish a persistent infection associated with the integration of viral nucleic acids in the host genome. Such persistence follows infection with a range of DNA viruses in the herpesvirus, adenovirus, and hepadenavirus families and RNA viruses in the retrovirus family. In the latter case, RNA is converted into DNA by the viral encoded reverse transcriptase prior to genomic integration (21,22). Other RNA viruses, such as measles virus, can also establish persistence within the central nervous system; however, the specific mechanisms which determine the occurrence of persistence have not been fully elucidated (23).
Viruses may also induce persistent pathological effects on the host after the virus has been cleared by the immune system. One mechanism for long-lasting effects is the generation of autoimmune interactions. Thus viral infection with the picornaviruses of the coxsackievirus family can induce an autoimmune myocarditis which can persist after the virus is cleared from cardiac tissues (24). Similarly, influenza virus infection can lead to the generation of antibodies which recognize brain proteins and which may initiate autoimmune phenomena within the central nervous system (25). It is of note, in this regard, that recent studies of schizophrenia have found evidence of autoimmune interactions as evidenced by anti-self antibodies and abnormal levels of cytokines within the central nervous system (26).
Infections with viruses and other agents can also induce persistent effect on the host by the destruction or alteration of cells crucial for normal functioning. One mechanism for this is the process of apoptosis or programmed cell death. Apoptosis is induced by a specific set of interactions between viral and cellular proteins, and probably reflects an attempt by the host to eliminate infect cells in a controlled manner. Apoptosis has been documented to be one of the central mechanisms involved in brain cell destruction following infection with herpes simplex virus and human immunodeficiency virus (27). Similarly influenza virus and other viruses have been documented to cause apoptosis in a subset of infected cells (28). In the case of infections with these agents, it is possible that the death of neural cells involved in critical signaling pathways within the central nervous system can lead to specific neurological and behavioral symptoms later in life. Due to the developmental nature of the maturation process within the central nervous system, the effect of cellular elimination might not be manifested until late in life when the affected cells become recruited for specific neural functions. It should be noted that, in such cases, viral antigens and nucleic acids derived from viruses which induce schizophrenia might not be detectable in the brain or cerebrospinal fluids of individuals when the symptoms first occur since they would have been cleared by the host immune system and the apoptotic process.
As noted above, viral antibodies have been more frequently detected in individuals with schizophrenia than have viral antigens or nucleic acids. This discrepancy may be the result of the long lag time between infection and the manifestation of disease. Since antibodies persist for a much longer period of time than antigens of nucleic acids, the detection of such antibodies may be a reflection of past infection. However, caution needs to be applied in the interpretation of antibody studies, even when carefully constructed control groups are employed in the analyses. The need for caution derives from the fact that some infectious and autoimmune diseases can result in polyclonal B-cell activation with the subsequent secretion of antibodies directed at a range of infectious and host-derived antigens. For example, infection with Epstein-Barr virus can result in the development of antibodies to a number of other viruses including measles, rubella, adenoviruses, enteroviruses and varicella-zoster virus (29). Similarly, infection with human immunodeficiency virus results in the development or augmentation of antibodies to a range of viral antigens as well as to host-derived antigens such as DNA, myosin, and ovalbumin (30). It is thus possible that the detection of antibodies to a range of viral agents may reflect infection with a more limited repertoire of infectious agents. Similarly, the presence of antibodies to host-derived proteins, noted in previous studies of schizophrenia (31), may reflect infected cells, as well as autoimmune pathogenic mechanisms. These considerations underscore the importance of prospective studies directed at determining the relationship between the occurrence of infection, the generation of the host immune response, and the development of clinical symptoms. Prospective studies could establish the temporal relationship of these events and directly determine the contribution of infection and immunity to disease pathogenesis. The early identification of individuals at risk for schizophrenia would markedly enhance the practicality and utility of such prospective studies.
Interaction of Infection with Genetic Factors
Much of the recent research in the area of schizophrenia has focused on genetic determinants of this disease. Factors stimulating investigations in this area include a higher than expected rate of schizophrenia in the first-degree relatives of affected individuals and an even higher rate of concordance in monozygotic twins (2). In addition, twin-adoption studies have indicated that the risk for schizophrenia correlates with the occurrence of this disease in the birth, but not the adoptive family of the adopted individual (32). However, it should be noted that the occurrence of genetic risk does not exclude a role for infectious agents in the etiology of schizophrenia. On the contrary, it is becoming increasingly recognized that the clinical course and outcome of most infectious diseases are dependent, to a large extent, on the interaction of the infecting microorganism with a range of host factors. While some aspects of the host response to infectious agents are determined by environmental factors such as nutrition and co-infection, others are clearly under genetic control. The most direct interactions between genetics and infection is evident in the function of the immune system. Severe defects in immune components are associated with frequent and often overwhelming infections caused by a variety of opportunistic pathogens. Since individuals with schizophrenia do not generally display evidence of opportunistic or overwhelming infections, it is unlikely that such genetic defects are directly involved in disease pathogenesis. However, genetic factors have been associated with more subtle responses to specific infectious agents. For example, susceptibility to Mycobacterium tuberculosis has been mapped to the Bcg gene localized in the short arm of human chromosome 2 between map positions q31 and q37 (33). This locus may also be involved in susceptibility to systemic mycobacteria such as Mycobacterium chelonei and Mycobacterium avium intracellulare (34). Susceptibility to other mycobacteria, such as Mycobacterium leprae, the causative agent of leprosy, is also under apparently genetic control associated with other loci (35). A human genetic susceptibility locus has also been identified for infection with the parasite Schistosoma mansoni, although the precise genomic localization of this susceptibility has not been determined (36).
If it is not eliminated by the host immune system, a pathogenic virus has to attach to and invade host cells. Many viruses and other intracellular microorganisms employ naturally occurring host proteins as sites of attachment and cellular entry. Genetic differences in the composition or quantity of such proteins can alter the host response to the infectious process. For example, the retrovirus Rous sarcoma virus employs the low-density lipoprotein (LDL) receptor as the site of cell binding and entry (37). Since this receptor is genetically diverse in human and animal populations (38), infection with viruses which use this receptor would be expected to display a range of infection efficiencies determined by the genetic makeup of the LDL receptor. Similarly, there is evidence that susceptibility to HIV-1 is determined by genetic variation in the composition of the CD4 receptor as well as by the quantity and structure of co-receptor molecules (39). A genetic susceptibility to the other human retrovirus HTLV-1 has also been documented, but the mechanism for this variation has not been determined (40). Genetic differences in receptor structure or function have also been postulated to determine host susceptibility to human rotavirus (41) and poliovirus (42) as well as to the bacteria Helicobacter pylori (43).
After a virus has bound to the cell membrane, additional cellular proteins are often required for viral internalization and transport within the cell as well as for the subsequent processing of viral peptides. For example, cellular proteins have been identified which specifically bind to influenza virus proteins following infection and which determine the occurrence of programmed cell death (44,45). Genetic variation in the composition or expression of these proteins provides another mechanism by which cellular proteins can determine the outcome of viral infection.
Genetic factors controlling response to infection may be particularly evident in the transmission of infection from mothers to their infants. In such cases, the mother transmits, not only the infecting agent, but also approximately half of the genetic components involved in host response (more in the case of host factors encoded by X-chromosomal or mitochondrial genes). Such virus-host interactions are likely to be operative in the efficient perinatal transmission of known agents such as cytomegalovirus, rubella, and human immunodeficiency virus as well as perinatally transmissible agents which have not yet been defined (46). In this context, it is possible that the association between schizophrenia in an adopted child and his or her birth parent may be partly explained by the acquisition of infection from the natural mother as well as by genetic factors. Similarly, the concordance rate for schizophrenia in monozygotic twins is similar to twin concordance rate of infectious diseases occurring during pregnancy or early life, suggesting that this concordance may be partly explained by the transmission of infection from the mother to one of her infants (2).
Models for Interactions between Infection and Host Factors
As noted above, the outcome of an infectious disease is determined, not only by the infecting microorganism, but also by the interaction of the organisms with the host. There are several models which can be employed for the analysis of this interaction between microbial and host factors. In all cases, the infecting agent invades the host and comes into contact with the host immune system. The ultimate outcome of the infectious process depends upon the pathogenic potential of the infecting agents and the nature of the immune response to infection.
In some cases, as depicted in the model in figure 1, the pathogenicity of the organism is so high that virtually all infected individuals develop some manifestation of infection.
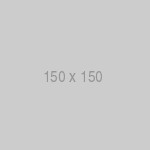
Fig. 1. Schematic model of infectious diseases with direct cause-and-effect relationships between the
initial infection and the ensuing disease. In such cases, virtually all infected individuals develop signs or symptoms of the disease.
This pattern is typical of the highly infectious agents in the human environment such as measles, cholera, varicella, smallpox, and plague. These organisms are responsible for large outbreaks in susceptible individuals. Fortunately, the incidence of many of these infections have been lowered by immunization and other programs directed at the protection of the human population. For this reason, many infectious diseases which now occur in human populations display more variable courses in the host (fig. 2).
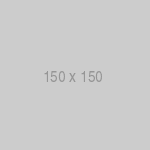
Fig 2: Schematic model of infectious diseases in which host factors play a major role in disease pathogenesis. In such cases, the manifestation of the disease is determined by the interaction between the pathogenicity of the infecting organism and the host reaction to infection. Thus many infected individuals may not develop disease manifestations following infection.
For example, an individual exposed to M. tuberculosis may develop no apparent symptoms, symptoms limited to respiratory tract, or disseminated disease associated with replication within the central nervous system or another organ. A similar variation in disease course is noted following infection with a large number of viral agents including herpesviruses as well as respiratory and gastrointestinal viruses. The disease course undergone by the infected individual is dependent upon a number of factors including the pathogenic properties of the organism as well as host factors relating to the immune status of the individual. As noted above, the immune status of an individual is determined by gene encoding proteins essential to the immune response as well as environmental factors, such as nutrition, which can alter the expression of these proteins.
Some human disease processes can be initiated by either infectious or noninfectious cases (fig. 3).
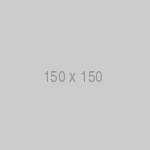
Fig. 3. Schematic model of infectious diseases in which infection represents only one of several possible disease etiologies. In such cases, the effect of infection can be mimicked by other environmental agents which interact with host factors in the ensuing disease process. In such cases, the relationship between infection and disease can be difficult to define using standard microbiological and environmental analytic methods.
In such cases, the disease process represents a final common pathway between tissue jury initiated by the infectious agent or other toxic environmental factors. For example, gastric ulcers can be caused by infection with the bacterium H. pylori or by medications such as corticosteroids or salicylates. The disease process is identical in both cases and the cause cannot be determined by pathological examinations; specific microbiological assays are required to determine the disease etiology (43). Similarly, the symptoms of gastroenteritis or hepatitis can be induced by either infectious or toxic agents; distinct infectious agents can interact with host factors to produce similar pathological processes. Interactions between genetic and infectious factors have also been postulated for a number of chronic diseases including insulin-dependent diabetes mellitus (47), postviral myocarditis (24), retroviral-induced bone disease (48), and neoplasia following infection with hepatitis B (49) and Epstein-Barr virus (50). In these cases, infections can cause chronic diseases similar to ones generated by other environmental factors. Such diseases are not ‘infectious’ in the classical sense in that they are not associated with the shedding of transmissible agents and they generally cannot be analyzed by standard microbiological and epidemiological methods. However, an understanding of the role of past or persistent infection in these diseases may lead to new methods for disease prevention and treatment.
It is likely that schizophrenia, if it is found to be associated with an infection, will fit into this category and that the understanding of the disease will require the elucidation of its microbial, genetic and environmental components. An important consequence of the elucidation of the role for infectious agents in the pathogenesis of schizophrenia is thus the possible application of anti-infectious agents to the treatment of this disease. It is of note in this regard that we have recently found that metabolites of the atypical neuroleptic drug clozapine are potent inhibitors of the in vitro replication of human retroviruses (50). In this context, the administration of an antimicrobial or antiviral agent can be considered to be a form of ‘gene therapy’ in the sense that the agents target the genome of the infecting microorganism. Despite advances in the development of methods for the regulation of human gene expression, it can be expected that the replication of viral and microbial nucleic acids will remain more amendable to pharmacological intervention than the corresponding replication and expression of human genes. The identification of microbial agents involved in the pathogenesis of some cases of schizophrenia would thus have a major impact on the development of new modalities for the treatment of this disease. Furthermore, since infections caused by many microbial pathogens are preventable by means of immunization and other immunoprophylactic measures, the elucidation of a role of such agents in the pathogenesis of schizophrenia would provide opportunities for the development of methods for the prevention of this disease in the human population.
This work was supported by the Stanley Foundation. The authors thank Dr. Christopher Ross for his careful reading of the manuscript.
References:
- Torrey EF: Surviving Schizophrenia, ed. 3. New York, Harpers Collins, 1995.
- Torrey EF, Bowler AE, Taylor EH, Gottesman II: Schizophrenia and Manic-Depressive Disorder. New York, Basic Books, 1994.
- Yolken RH, Torrey EF: Viruses, schizophrenia, and bipolar disorder. Clin Microbiol Rev 1995;8:131-145.
- O’Callaghan E, Gibson T, Colohan HA, Walshe D, Buckley P, Larkin C, Waddington J: Season of birth in schizophrenia; evidence for confinement of an excess of winter births to patients without a family history of mental disorder. Br J Psychiatry 1991;158:764-769.
- Mednick SA, Machon RA, Huttunen MO, Bonett D: Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry 1988;45:189-192.
- Adams W, Kendell RE, Hare EH, Munk-Jergensen P: Epidemiological evidence that maternal influenza contributes to the aetiology of schizophrenia: An analysis of Scottish, English and Danish data. Br J Psychiatry 1993;163:522-534.
- Barr CE, Mednick SA, Munk-Jergenson P: Exposure to influenza epidemics during gestation and adult schizophrenia: A 40-year study. Arch Gen Psychiatry 1990;47:869-874.
- Sham PC, O’Callaghan E, Takei N, Murray GK, Hare EH, Murray RM: Schizophrenia following pre-natal exposure to influenza epidemics between 1939 and 1960. Br J Psychiatry 1992;160:461-466.
- Torrey EF, Rawlings R, Waldman IN: Schizophrenic births and viral diseases in two states. Schizophr Res 1988;1:72-77.
- Sham PC, Maclean CJ, Kendler KS: Risk of schizophrenia and age difference with older siblings: Evidence for a maternal viral infection hypothesis? Br J Psychiatry 1993;163:627-633.
- Eagles JM, Gibson I, Bremner MH, Clunie F, Ebmeier KP, Smith NC: Obstetric complications in DSM-III schizophrenics and their siblings. Lancet 1990;335:1139-1141.
- Lewis SW, Murray RM: Obstetric complications, neurodevelopmental deviance and risk of schizophrenia. J Psychiatr Res 1987;21:413-421.
- Pandurangi AK, Pelonero AL, Nadel L, Calabrese VP: Brain structure changes in schizophrenics with high serum titers of antibodies to herpes virus. Schizophr Res 1994;11:245-450.
- Monhammed AK, Magnusson O, Maehlen J, Fonnum F, Norrby E, Schultzberg M, Kristensson K: Behavioural deficits and serotonin depletion in adult rats after transient nasal viral infection. Neuroscience 1990;35:355-363.
- Zimmermann W, Dô rrwald R, Ludwig H: Detection of Borna disease virus RNA in naturally infected animals by a nested polymerase chain reaction. J Virol Methods 1994;46:133-143.
- Griffin DE, Hardwick JM: Neurovirulent strains of alphavirus induce apoptosis in bcl-2-expressing cells: Role of a single amino acid change in the E2 glycoprotein. Proc Natl Acad Sci USA 1994;91:5205-5206.
- Wohrmann T, Hewicker-Trautwein M, Fernandez A, Moenning v, Liess B, Trautwein G: Distribution of bovine virus diarrhea viral antigens in the central nervous system of cattle with various congenital manifestations. Zentralbl Veterinarmed 1992;39:599-609.
- Kaufmann CA, Weinberger DR, Stevens JR, Asher DM Kleinman JE, Sulima MP, Gibbs CJ, Gadjusek DC: Intracerebral inoculation of experimental animals with brain tissue from patients with schizophrenia: Failure to observe consistent or specific behavior and neuropathological effects. Arch Gen Psychiatry 1988;45:648-652.
- Parnas J, Cannon TD, Jacobsen B, Schulsinger H, Schulsinger F, Mednick SA: Lifetime DSM-III-R diagnostic outcomes in the offspring of schizophrenic mothers. Results from the Copenhagen high-risk study. Arch Gen Psychiatry 1993;50:707-714.
- Coutlee F, Viscidi RP, Saint-Antoine P, Kessous A, Yolken RH: The polymerase chain reaction: A new tool for the understanding and diagnosis of HIV-1 infection at the molecular level. Mol Cell Probes 1991;5:241-259.
- Katz RA, Shalka AM: Generation of diversity in retroviruses. Annu Rev Genet 1990;24:409-415.
- Sablitzky F, Jonsson J-I, Cohen BL, Phillips RA: High-frequency expression of integrated proviruses derived from enhancer trap retroviruses. Cell Growth Differ 1993:4:451-459.
- Atloms GK, Balluz IM, Glasgow GM, Mabruk MJ, Natale VA, Smyth JM, Sheahan BJ: Analysis of the molecular basis of neuropathogenesis of RNA viruses in experimental animals: Relevance for human disease? Neuropathol Appl Neurobiol 1994;20:91-102.
- Lane JR, Neumann DA, Lafond-Walker A, Herskowitz A, Rose NR: Role of IL-1 and tumor necrosis factor in coxsackievirus-induced autoimmune myocarditis. J Immunol 1993;151:1682-1690.
- Laing P, Knight JG, Hill JM, Harris AG, Oxford JS, Webster RG, Markwell MAK, Paul SM, Pert CT: Influenza viruses induce autoantibodies to a brain-specific 37-kD protein in rabbit. Proc Natl Acad Sci USA 1989;86:1998-2002.
- Sirota P, Schild K, Elizur A, Djaldetti M, Fishman P: Increased interleukin-1 and interleukin-3-like activity in schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry 1995;19:775-783.
- Geiger KD, Gurushanthaiah D, Howes EL, Lewandowski GA, Reed JC, Bloom FE, Sarvetnick NE: Cytokine-mediated survival from lethal herpes simplex virus infection: Role of programmed neuronal death. Proc Natl Acad Sci USA 1995;92:3411-3415.
- Takizawa T, Fukuda R, Miyawaki T, Ohashi K, Nakanishi Y: Activation of the apoptotic Fas antigen-encoding gene upon influenza virus infection involving spontaneously produced beta-interferon. Virology 1995;209:288-296.
- Haukenes G, Viggen B, Boye B, Kalvenes MB, Flo R, Kalland KH: Viral antibodies in infectious mononucleosis. FEMS Immunol Med Microbiol 1994;8:218-224.
- Shirai A, Cosentino M, Leitman-Klinman SF, Klinman DM: Human immunodeficiency virus infection induces both polyclonal and virus-specific B cell activation. J Clin Invest 1992;89:561-566.
- Yang ZW, Chengappa KNR, Shurin G, Brar JS, Rabin BS, Gubbi AV, Ganguli R: An association between anti-hippocampal antibody concentration and lymphocyte production of IL-2 in patients with schizophrenia. Psychol Med 1994;24:449-455.
- Tienari PJ, Wynne LC: Adoption studies of schizophrenia. Ann Med 1994;26:233-237.
- Skamene E: The Bcg gene story. Immunology 1994;191:451-460.
- Levin M, Newport MJ, D’Souza S, Kalabalikis P, Brown, IN, Lenicker HM, Agius PV, Davies EG, Thrasher A, Klein N, et al: Familial disseminated atypical mycobacterial infection in childhood: A human mycobacterial susceptibility gene? Lancet 1995;345:79-83.
- Levee G, Liu J, Gicquel B, Chanteu S, Schurr E: Genetic control of susceptibility to leprosy in French Polynesia: No evidence for linkage with markers on telemeric human chromosome 2. Int J Lepr Other Mycobac Dis 1994;62:499-511.
- Levee G, Liu J, Abel L, Demenais F, Prata A, Souza AE, Dressein A: Evidence for the segregation of a major gene in human susceptibility/resistance to infection by Schistosoma mansoni. Am J Hum Genet 1991;48:959-970.
- Bates P, Young JA, Varmus HE: A receptor for subgroup A Rous sarcoma virus in related to the low density lipoprotein receptor. Cell 1993;74:1043-1051.
- Herz J, Willnow TE: Lipoprotein and receptor interactions in vivo. Curr Opin Lipidol 1995;6:97-103.
- Williams LLM, Cloyd MW: Polymorphic human gene(s) determines differential susceptibility of CD4 lymphocytes to infection by certain HIV-1 isolates. Virology 1991;184:723-728.
- Okayama A, Chen YM, Tachibana N, Shiori S, Lee TH, Tsuda K Essex M: High incidence of antibodies to HTLV-1 tax in blood relatives of adult T cell leukemia patients. J Infect Dis 1991;163:47-52.
- Willoughby RE, Yolken RH: SA11 rotavirus is specifically inhibited by an acetylated sialic acid. J Infect Dis 1990;161:116-119.
- Nomoto A, Koike S, Aoki J: Tissue tropism and species specificity of poliovirus infection. Trends Microbiol 1994;2:47-51.
- Go MF, Graham DY: How does Helicobacter pylori cause duodenal ulcer disease:The bug, the host, or both? J Gastroenterol Hepatol 1994;9:S8-S10.
- O’Neil RE, Palese P: NPI-1, the human homolog of SRP-1, interacts with influenza virus nucleoprotein. Virology 1995;206:116-125.
- Takizawa T, Fukuda R, Miyawaki T, Ohashi K, Nakanishi Y: Activation of the apoptotic Fas antigen-encoding gene upon influenza virus infection involving spontaneously produced beta-interferon. Virology 1995;209:288-296.
- Anteby EY, Yagel S: Immune responses to viral infection; in Gonik B (ed): Viral Diseases in Pregnancy. New York, Springer 1994, pp. 1-11.
- Fohlman J, Friman G: Is juvenile diabetes a viral disease? Ann Med 1993;25:569-574.
- Labat ML: A new approach to the study of the origin of genetic diseases: Retroviral etiology of osteopetrosis. Biomed Pharmacother 1991;45:23-27.
- Shen FM, Lee MK, Gong HM, Cai XG, King MC: Complex segregation of analysis of primary hepatocellular carcinoma in Chinese families: Interaction of inherited susceptibility and hepatitis B viral infection. Am J Hum Genet 1991;49:88-93.
- Raab-Traub N: Epstein-Barr virus infection in nasopharyngeal carcinoma. Infect Agents Dis 1992;1:173-184.